
Screenshot of Allegory Magazine’s Archives: Volume 45/72 (Spring/Summer 2024)


I have compiled this list to help myself and others learn about the entrepreneurial landscape and find opportunities for funding. Although experience is the best teacher, these resources can act as a foundation for exploring further avenues of learning. I should note that many (but not all) of these resources are aimed at academic researchers seeking to spin off companies based on new biotechnology inventions. The resources are listed in alphabetical order.
Academic Entrepreneurship How to Bring Your Scientific Discovery to a Successful Commercial Product: book aimed at educating academic entrepreneurs which details important topics like IP, funding, market research, and more. “The pathway to bringing laboratory discoveries to market is poorly understood and generally new to many academics. This book serves as an easy-to-read roadmap for translating technology to a product launch – guiding university faculty and graduate students on launching a start-up company”.
Activate Fellowship: a 2-year fellowship program (full time) for scientists working on startups. Provides guidance on business development, networking, $100K in funding for R&D, $100K per year living stiped, and technical support. “Activate transforms scientists and engineers into founders, empowering them to reinvent the world by bringing their research to market. The two-year Activate Fellowship provides early-stage science entrepreneurs with funding, technical resources, and unparalleled support from a network of scientists, engineers, investors, commercial partners, and fellow entrepreneurs”.
America’s Seed Fund: official website of the U.S. government’s SBIR (Small Business Innovation Research) and STTR (Small Business Technology Transfer) grants. Directs to many more specific resources on how to apply for these grants, what governmental agencies offer them, etc. “America’s Seed Fund awards non-dilutive funding to develop your technology and chart a path toward commercialization. The federal government invests in your solution and gives you the freedom to run your business according to your vision”.
NIH SEED: official website of the NIH’s SBIR and STTR programs which offers resources on funding opportunities, how to apply, entrepreneurial training, etc. “SBIR and STTR grant funding opportunities offer small business entrepreneurs a chance to obtain non-dilutive funding for early-stage research and development. Applications are accepted three times a year… NIH advertises the availability of grant support through notices of funding opportunities (NOFOs), previously referred to as funding opportunity announcements (FOAs)… Researcher-initiated ideas are proposed via the SBIR and STTR Omnibus grant solicitations. These funding opportunities do not specify a topic, though they link to identified topics of interest for each participating awarding component. Most small business applications to NIH are submitted to the Omnibus solicitations”.
Arch Grants: a source of up to $100,000 of funding for St. Louis startup companies which is awarded through an annual startup competition. Improved access to an ecosystem of resources for business development is also provided to winners. “Through our unique and groundbreaking annual Startup Competition model, we provide up to $100,000 in equity-free grants and access to an ecosystem of resources, helping early-stage startups grow and scale”.
WashU Venture Network Follow-on Investments: a funding program of up to $150,000 for companies with ties to Washington University in St. Louis that are also previous Arch Grants winners. “The WashU Venture Network Follow-on Investments is a joint effort between WashU’s Skandalaris Center for Interdisciplinary Innovation and Entrepreneurship and the ‘In St. Louis, For St. Louis’ initiative. It will award up to $150,000 each year to companies with WashU ties that previously have been awarded funding through Arch Grants, the local nonprofit that awards equity-free grants to startups”.
Cake Equity SAFE Notes: a guide from Cake Equity on what a SAFE (Simple Agreement for Future Equity) is and how it works. “What is a SAFE note agreement, how does it work, how is it different from convertible notes, and how to leverage it to raise funds for your startup”.
Charles River: a high-end contract research organization which provides a wide array of services for biotechnology R&D. “Charles River provides essential products and services to help pharmaceutical and biotechnology companies, government agencies, and leading academic institutions around the globe accelerate their research and drug development efforts. We deliver what our clients need to improve and expedite the discovery, early-stage development, and safe manufacture of new therapies for patients who need them”.
Charles River AAV Testing: a part of Charles River’s contract research organization which provides GMP characterization and in vitro AAV testing services for gene therapy R&D. “Adeno-associated virus (AAV) is one of the most widely used vectors in gene therapy. Our extensive, in-house AAV testing experience with rigorous characterization and biosafety testing will help ensure that your gene therapy is safe, effective, and regulatory-compliant from preclinical stages to commercialization”.
Charles River Cell Banking: a part of Charles River’s contract research organization which provides cell banking, which can facilitate consistent production biologics for clinical applications. “Cell banking is the foundation of biologics and therapeutics. With decades of experience and over 2,000 cell and/or viral banks successfully produced, our fully integrated CGMP cell bank production, storage, and characterization services will help support your product’s success from R&D through commercial manufacturing”.
Charles River CRADL Vivarium Lab Space: a part of Charles River’s contract research organization which provides animal testing services for biomedical R&D. “Charles River’s Accelerator and Development Lab, provides contract vivarium lab space with built-in animal husbandry, veterinary care, operations management, and IACUC protocol support to give you greater speed, flexibility, and control over your mice and rat in vivo pharmacology studies”.
Charles River GMP AAV Production Service: a part of Charles River’s contract research organization which provides GMP-grade AAV manufacturing services. “As a trusted CDMO partner for AAV production services, our multi-disciplinary teams provide the experience and dedication to quality that you need to scale up your AAV gene therapy project into a patient-ready product. Leverage the nAAVigation® AAV manufacturing platform to bolster your program with all-important predictability, guidance, and reduced time to clinic”.
Cooley GO: a website with articles about business concepts for entrepreneurs at various stages of developing startups. “At Cooley, we want to give you the information you need to build a great company – easily accessible and from the most trusted source”.
Cooley GO: What You Should Know About SAFEs: an article from Cooley GO on the SAFE (Simple Agreement for Future Equity) investment mechanism. “A SAFE is an investment instrument that converts the holder’s value into equity of the issuer upon certain triggering events”.
Curie.Bio: a therapeutics-focused VC firm that provides funding at the seed and series A stages as well as extensive guidance and assistance to scientific founders building biomedical companies. They offer a ‘co-pilot’ program to guide founders towards optimal therapeutic targets, a network of CROs, and more. Has a low acceptance rate (~1%) for funding. “We built Curie.Bio to free the founders. Curie.Bio significantly improves your ability to create impactful medicines while minimizing long-term dilution. Our unparalleled team and capital-efficient model provide you with immediate access to the industry’s top drug hunters, operators, and R&D services from day one”.
Emergent Ventures Grant: a philanthropic funding opportunity offered to highly motivated entrepreneurs (and others) with scalable project ideas for meaningfully improving society. Most awardees receive around $10K-$20K, but there are rumors that larger awards have been issued as well. Has a simple online application process centering on a 1500-word proposal. “Launched in 2018, Emergent Ventures is a low-overhead fellowship and grant program that supports entrepreneurs and brilliant minds with highly scalable, ‘zero to one’ ideas for meaningfully improving society. Mercatus Center faculty director Tyler Cowen administers the program”.
Experiment: a crowdfunding organization that provides small grants to scientific projects if donations meet a user-defined goal amount. “Experiment is an online platform for discovering, funding, and sharing scientific research… Creators never give up any ownership of their work to Experiment or backers. You keep 100% ownership and control over your work”.
Fifty Years: a VC firm focused on funding early-stage deep tech companies with technical founders. They also provide guidance to help technical/scientific founders build companies. “Fifty Years is a pre-seed and seed focused VC firm. We back founders using technology to solve the world’s biggest problems. We also help start companies. 50Y founders are building massive businesses while solving the world’s most important problems: the climate crisis, disease, connectivity, malnutrition, and more”.
Spinout Playbook: a guide from Fifty Years which provides advice for spinning companies out from inventions in academic laboratories. “We’ve seen negotiations with tech transfer offices (TTOs) take so long that founders burned out, teams lost momentum, and investors walked away. We’ve seen TTOs introduce terms that hurt the startup’s ability to raise capital. We’ve seen so many exotic term combinations that we wondered: how are founders supposed to make sense of it all? So we made this Spinout Paybook to help aspiring scientist founders make sense of the process”.
Manifest Grants: a fast grants program run by Fifty Years which offers pre-seed funding to scientists working on translational ideas. Applications are closed as of 2/2/2025 but may reopen at some point. “Manifest Grants is a fast grants program that awards $25K – $100K to scientists to accelerate their most ambitious ideas to solve the world’s biggest problems. We focus on translational research. The applications are max 2 pages and you get a decision within 21 days. No IP is taken. Are you an academic scientist working on a translational research idea for solving a big problem? Follow us on X (Twitter) to keep an eye out for updates on our grants programs. Applications are closed at the moment”.
Hevolution Foundation: a Saudi Arabian organization with a mission to treat aging and improve healthspan. It has an annual budget of $1B and provides a variety of grants and opportunities (for both translationally minded academics and companies). “Hevolution Foundation is the first non-profit to pursue age-related therapeutic breakthroughs with a commitment to funding global scientific discovery, and investing in private companies and entrepreneurs who are dedicated to advancing aging science. Through the acceleration of science, we can decelerate aging and its consequences”.
How to Start a Life Science Company: A Comprehensive Guide for First-Time Entrepreneurs: a book which provides a helpful primer on important steps for forming, growing, and running life sciences companies. “This comprehensive guide takes first-time entrepreneurs through every step of founding a life science company. It covers all the business basics that we aren’t taught in science and engineering courses”.
How to Build a Biotech: part of Celine Halioua’s blog where she provides educational writeups on a variety of topics for biotechnology entrepreneurship. “The Summer of 2019, I (Celine) gave a series of lectures to Longevity Fund’s Venture Fellows on the basics of building a biotechnology company. This is the write up of those lectures, with some additions by Laura Deming & myself (Celine)”.
Basics of Life Science Patents: great educational blog post by Celine Halioua providing an introduction to what one should know about IP protection for biotechnology. “There are three broad categories of patents: utility, plant, and design patents. Generally, biotech patents are utility patents, like the vast majority (>90%) of patents. Utility patents are enforceable for 20 years, beginning from date of earliest filing. This ticker starts when you file a provisional patent, despite the fact that the patent may not be granted for years after this filing”.
Venture Capital for Bio 101: excellent educational blog post by Laura Deming and Celine Halioua which explains the process, challenges, and strategies of seeking VC funding for biotechnology startups. “Venture capitalists are investors who specifically invest in high risk, high reward companies. Venture capitalists invest capital in your company in exchange for equity, in the hope that the equity is worth significantly more in the future. This post will cover how to find and convince such a person to swap money for equity in your new company”.
Investopedia: an online encyclopedia with numerous detailed articles covering topics in business, entrepreneurship, and investing. “Investopedia was founded in 1999 with the mission of helping people improve their financial outcomes”.
Common Stock: What It Is, Different Types, vs. Preferred Stock (Investopedia article): an article on Investopedia that discusses the concepts of common stock and preferred stock. “Common stock is not just a piece of paper—or, these days, a digital entry—but a ticket to ownership in a company. When you hold common stock, you get to weigh in on corporate decisions by voting for the board of directors and corporate policies”.
Par Value of Stocks and Bonds Explained (Investopedia article): an article on Investopedia that discusses the concept of par value for stocks and bonds. “Par value, also known as nominal or original value, is the face value of a bond or the value of a stock certificate, as stated in the corporate charter. Par value is required for a bond or a fixed-income instrument and shows its maturity value and the dollar value of the coupon, or interest, payments due to the bondholder”.
J.P. Morgan Healthcare Conference: an important event where biomedical entrepreneurs network, negotiate, and make deals with investors as well as form other kinds of partnerships. “This premier conference is the largest and most informative health care investment symposium in the industry which connects global industry leaders, emerging fast-growth companies, innovative technology creators and members of the investment community”.
LegalZoom: offers services to help handle the legal paperwork associated with starting and maintaining a business. “A tech company dedicated to making legal help accessible to all… launched 10 online services, focusing on estate planning, business formation, and intellectual property protection… easier, and less expensive way to get legal help… an independent network of attorneys so our customers could get personalized legal advice – without having to leave home”.
Business formation: offers packages for helping to incorporate businesses and work through the legal process of doing so. “Choose your business type… Answer a few questions… We’ll complete and file your paperwork”.
List of Biotechnology Companies to Watch: my own list of innovative biotechnology companies that are making an impact. Includes links to the company websites as well as key facts about each. May be useful for exploring landscapes of competitors and potential partners. “I created this list of organizations… to serve as a resource to help people learn about and keep track of key biotechnology companies. Some of these are emerging startups, some are established giants, and some provide useful services. Some notable nonprofit organizations are included as well”.
LongeVC Venture Fellowship Program: a remote 6-month program that guides ambitious graduate students and postdocs through learning about business strategy. “This program offers a unique opportunity to collaborate closely with our investment team, assisting in evaluating potential investments, participating in company pitches, and developing strategic initiatives. Fellows will also have the chance to network with industry experts and gain exposure to cutting-edge fields such as drug discovery and development, computational and synthetic biology, and genomic medicine. This is a part-time, remote, 6-month paid fellowship, with the potential for extension based on performance and mutual interest”.
NSF I-Corps: a program from the U.S. government that helps guide small businesses and make them more successful. Teams can apply for the program and then will be taken through an intensive experiential learning program to develop their company. “Launched to support NSF’s mission through experiential learning using the customer discovery process – allowing teams to quickly assess their inventions’ market potential. I-Corps prepares scientists and engineers to extend their focus beyond the laboratory to increase the economic and societal impact of NSF-funded and other basic research projects”.
I-Corps hubs: a program wherein I-Corps provides local training at certain locations and in partnership with certain institutions to help new startup teams learn about entrepreneurship. “Bring together institutions of higher education within a distinct geographical region to collaborate and deliver a standardized curriculum. The curriculum explores the commercial potential of deep technologies with members of the scientific community and other interested parties in the Hub’s region”.
I-Corps teams: the program that I-Corps uses to guide early startup teams through the process of developing their business. “Supports teams of scientists and engineers to explore the commercial potential of technologies developed in university laboratories through a standardized entrepreneurial training program”.
Nucleate: a student-led organization which identifies, educates, connects, and empowers academic biotechnology entrepreneurs to create and run startups. George Church, Tom Kalil, and Pamela Silver are some of the advisors. “Nucleate is a student-led organization that represents the largest global community of bio-innovators”.
Nucleate Activator: a six-month entrepreneurship program that equips academic biotechnology founders with the skills and connections for successful business development. “Activator is a six-month, equity-free cohort program designed for academic biotech founders. Its curriculum has served more than 100 life sciences ventures that have raised over $190 million in venture funding. Participating teams refine their scientific discoveries into biotech venture theses and train under Nucleate’s unparalleled network, rigorous curriculum, funded fellowships, legal support, and highly subsidized perks. Activator culminates in a final pitch showcase before world-renowned judges”.
Nucleate Operationalizing Your Therapeutics Spinout Playbook: an educational booklet which provides guidelines for academic biotechnology entrepreneurs on how to create and operate startup companies. “Successfully transitioning academic research to real world applications is a significant challenge for life science entrepreneurs. The goal of this playbook is to provide a comprehensive guide for academic founders, covering essential aspects of company creation and operational strategies. In partnership with Alexandria LaunchLabs and Curie.Bio”.
Petri: a Boston venture firm which provides mentorship, resources, and funding for biotechnology founders. “Petri is co-founded by a group of leaders who have deep experience building ideas into iconic companies, and academics who are driving foundational science in bioengineering. We believe the future of biotech lies in supporting the next-generation of founders who will lead us there. Petri surrounds founders with the support they need to build world-class teams, grow as leaders, develop intellectual property, find customers, and raise capital”.
O’Shaughnessy Fellowship: a remote program that offers $100K to ten researchers, builders, and creatives to work on boldly innovative projects (it appears early-stage companies might be eligible, although the fellowship is primarily aimed at individuals). Also offers $10K plus networking opportunities via the O’Shaughnessy grants program to twenty more people working on exciting projects. “This is the O’Shaughnessy Fellowships & Grants. A one-year program that unites the world’s most bold and undiscovered researchers, builders and creatives to find, build and spread new ideas. In just two years, we’ve scoured over 140 countries to find and fund 55 extraordinary innovators, leading to six companies & non-profits, five documentaries, >2.2 billion views on YouTube, and the blossoming of numerous scientific, creative, and technological projects”.
O’Shaughnessy Infinite Adventures: the investment arm of O’Shaughnessy Ventures which offers funding, networking, and support. They are mostly looking for investment opportunities of $1M+, but they sometimes invest <$1M. “Conventional VC firms often only provide funding and some advice. We are a multidimensional investment company that not only offers capital, but also amplifies your story through our ever-growing media network”.
Pillar VC: a VC firm that focuses on backing transformative technology company founders at early stages. They have a particular focus on academic spinoff companies. Has a team of experienced CEOs to provide guidance to new founders. “The term VC itself evokes an evil empire — the dark side — a painful lack of alignment between investors and entrepreneurs. In a business where trust is paramount, the dark side created a world that put us at odds with those we needed to trust most. Pillar is fixing that”.
Pillar VC Moonshot: a startup competition for academic spinoffs which awards at least two winners $250K SAFEs and one winner a $1M SAFE. “At least one member of your Founding Team must be a university undergraduate, grad student, postdoc or faculty member. Whether you have an idea for an industry-leading company or a groundbreaking technology, all ideas are open for consideration”.
SciFounders: an investment group that funds founders who have technical backgrounds. Offers a fellowship that provides up to $1M as well as mentorship to technical founders working on innovative early-stage startups. “We back strong technical leaders who work on world-changing technologies from pre-seed to series A. We also run SciFounder Fellowship which comes with up to $1MM to get started and hands-on mentorship from us”.
Soma Capital: a non-traditional VC firm with a focus on disruptive technologies and highly driven founders, aims to be more founder friendly than other such firms. “Soma Capital invests in brilliant, fearless teams building Category Kings. We focus on software to automate the world, across any sector and geography that can touch billions of people and push humanity forward”.
Project 31: a non-dilutive pre-seed funding opportunity that Soma Capital offers to help kick off the beginnings of new startups. “An initiative by Soma Capital to empower today’s pioneers with $11,111, a vast network of mentors, and the infrastructure you need to materialize ambitions”.
Soma Fellows: a program that provides funding, mentorship, and network connections for driven entrepreneurs starting disruptive new tech companies. “Teams have the opportunity to receive $100k to 1m in funding from Soma Capital at the most founder-friendly terms possible. Soma Cap has the firepower to keep supporting you across the full path to IPO and the early stages are just the beginning for us… The program spans several weeks, offering sessions that guide you through the early stages of your entrepreneurial journey and help you build a foundation for scaling lasting companies. The Soma team, leveraging years of experience with top companies of our generation, will become your closest allies in establishing your company’s core and creating a product that impacts billions. The program culminates in a demo day, where founders showcase their products to potential customers, investors, and the community”.
So You Want to Start a Biotech Company: an article published in Nature Biotechnology which provides an overview of some of the key steps, challenges, and strategies needed for starting a biotechnology company. “Commercializing research is fraught with pitfalls, but a thoughtful checklist can ensure you set off on the right path and give your fledgling business the best chance of success”.
Thrive Capital: an investment firm focusing on technology companies including some biotechnology companies (esp. AI for biology). “Thrive Capital is an investment firm that builds and invests in internet, software, and technology-enabled companies”.
Trailblazer List: a resource which lists a variety of highly impactful companies across different fields. May be useful for exploring landscapes of competitors and potential partners. “There are endless meaningful problems. Join a team building the future. Make science-fiction real. Explore something different. Discover deep-tech companies”.
Veterinary Innovation Program (FDA): an FDA-hosted program that offers regulatory guidance (as well as enhanced review efficiency) to companies developing genetically engineered animal, tissue, or cell products. “The FDA Center for Veterinary Medicine’s (CVM) Veterinary Innovation Program (VIP) is for certain intentional genomic alterations (IGA) in animals and animal cells, tissues, and cell- or tissue-based products (ACTPs). The goal of the VIP is to facilitate advancements in development of innovative animal products by providing greater certainty in the regulatory process, encouraging development and research, and supporting an efficient and predictable pathway to market for ACTPs and IGAs in animals”.
Y Combinator Guide to Seed Fundraising: a publicly available set of recommendations from Y Combinator on concepts, terminology, and best practices for startup seed round fundraising from VCs, angel investors, etc. “This brief guide is a summary of what startup founders need to know about raising the seed funds critical to getting their company off the ground”.
Y Combinator SAFE documents: publicly available legal SAFE (Simple Agreement for Future Equity) documents used by Y Combinator. Several versions with different conditions are available. “Y Combinator introduced the safe (simple agreement for future equity) in late 2013, and since then, it has been used by almost all YC startups and countless non-YC startups as the main instrument for early-stage fundraising”.
Z Fellows: a startup fellowship which offers $10K at a $1B valuation cap (or participation without the money), mentorship and guidance, connections, and a 1-week workshop at the end of the program. Does not require fellows to relocate. “Z Fellows are technical builders of all ages working on side projects and startups. We are your first believer. We’ve worked with high school dropouts, college students, and people with full-time jobs across a variety of industries, including consumer, social, enterprise, defense, healthcare, edtech, fintech, gaming, cloud infrastructure, cybersecurity, crypto, AI, ML, climate, biotech, and more. Z Fellows is a one-week startup program. But it really doesn’t end after one week. We continue to help you for the life of your company, and beyond — and so does the ZF alumni community”.
50 Biotech Seed & Angel Investors to Check Out in 2024: an article listing 50 groups/organizations that invest in early-stage biotechnology companies and providing some information about each. “In this blog post, we’ll explore the landscape of biotech seed and angel investors in 2024, highlighting key players who are shaping the future of this dynamic industry”.
100 Plus Capital: an investment group that focuses on funding companies that could improve human longevity. “100 Plus Capital invests in companies positively impacting human longevity. This can be directly targeted (for example, anti-aging companies) or broader reaching (clean food and water companies). If you are working on extending human healthspan, we are interested in hearing your ideas”.
1517 Fund: a venture capital group that provides very early-stage funding to highly motivated and rebellious outside-the-box entrepreneurs, particularly young dropouts and sci-fi scientists working at the cutting edge of technology. “1517 is a venture capital fund and community supporting college dropouts solving hard problems and deep tech scientists with investment at the earliest stages of their companies. Founded by the cofounders of the Thiel Fellowship, it supports founders across software, hardware, and deep tech verticals and also provides a community to hackers, makers, and scientists from across the world”.
6 Steps to Incorporating Your Business: a simple webpage published by the U.S. Chamber of Commerce that provides an overview of the business incorporation process and options. “If you are considering incorporating your business, it simply means you are creating an entity that is legally separate from you. A corporation can own property and sign contracts, it can be sold and carry on without your involvement, and, if it goes bankrupt, you are not held personally liable. While every state handles the process a bit differently, there are six basic steps you should keep in mind”.

I’m writing these notes to help myself and others review mathematical probability (mostly from an applied perspective). So far, I have covered single-variable probability. I plan to later add sections on joint probability and perhaps a few other topics. My main source is the excellent textbook “Probability, Statistics, and Random Processes” by Hossein Pishro-Nik.
Axioms of probability
The axioms of probability include: (i) for any event A, P(A) ≥ 0, (ii) the probability of the total sample space S is P(S) = 1, and (iii) if A1,A2,A3,… are disjoint events then P(A1∪A2∪A3…) = P(A1) + P(A2) + P(A3)…
Note that intersection means “and” while union means “or”. Thus P(A∩B) = P(A and B) = P(A,B) while P(A∪B) = P(A or B).
An important tool in probability is the inclusion-exclusion principle. This is given by the following formulas for two events and for n events.

Conditional probability
Consider two events A and B in sample space S. The conditional probability of A given B is defined by the equation below.

Two events are independent if the first equation below holds true. Three events are independent if the second, third, fourth, and fifth equations below all hold true. This logic can be combinatorically extended to n independent events as well.

The law of total probability is an important tool for working with conditional probability. If B1,B2,B3,… represents a partition of the sample space S (the sample space can be “split” or partitioned into n “regions” or disjoint sets Bi), then the law of total probability is given as follows for any event A.

Bayes theorem represents an extremely important and widely consequential tool for conditional probability. Learning the intuition behind Bayes theorem can be highly valuable. The equation for Bayes theorem with two events A and B where P(A) ≠ 0 is given by the first equation below. The equation for Bayes theorem with n events B1, B2, B3… which partition the sample space S while P(A) ≠ 0 is given by the second equation below.
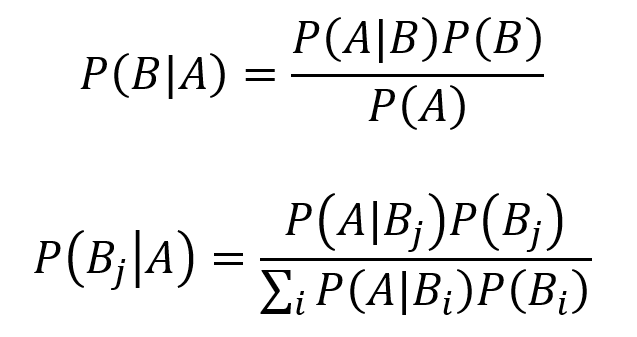
Two events are conditionally independent if they satisfy the following formula.

Random variables
A random variable X is a function mapping the sample space to the real numbers. That is, X is a real-valued function that assigns a value to each possible outcome of a random experiment.
The range of X is the set of possible values for X. As an example, flip a coin 5 times and define X as the number of heads that occur. The range of X is the set RX = {0,1,2,3,4,5}.
Discrete random variables are those which have countable ranges. Examples of countable ranges include finite sets as well as countably infinite sets like ℤ, ℚ, and ℕ.
Continuous random variables are those which have uncountable ranges. Any non-empty subset of ℝ is uncountable (as is the entirety of ℝ).
Probability mass functions
Probability mass functions (PMFs) assign probabilities to the possible values of a discrete random variable. If X is a discrete random variable with a finite or countably infinite range RX = {x1,x2,x3,…,xn}, then P(xk) = P(X = xk) for k = 1,2,3,…,n is the probability mass function of X.
As an example, the PMF for tossing a coin twice and setting X as the number of heads observed is as follows.

As it is a probability measure by definition, the PMF has the properties of a probability measure.
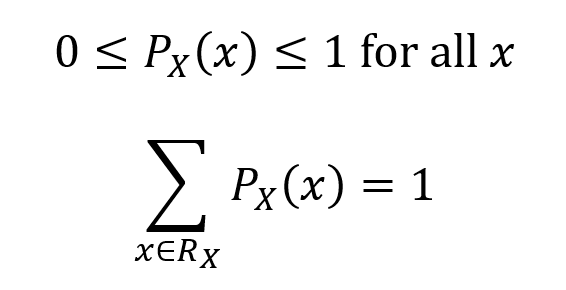
In addition, for any set A which is a subset of RX, the probability that X ∈ A is given as the sum of the probabilities of X for each element of A.

Independent random variables
Consider two discrete random variables X and Y. If the following equation holds true, then X and Y are independent.
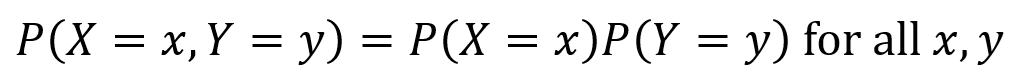
X and Y are furthermore independent if knowing the probability of one of them does not alter the probabilities of the other. Note the vertical line “|” denotes “given”, so P(X =x|Y=y) is probability that X=x given that Y=y.
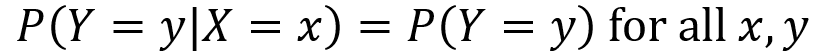
Special discrete distributions
Bernoulli distribution: a random variable X is a Bernoulli random variable with parameter p if its PMF is given by the first equation below. Bernoulli random variables indicate when a certain event A occurs, so they take on a value of 1 if A happens and 0 otherwise. This is described in the second equation below. Here, 0 < p < 1.

Geometric distribution: a random variable X is a geometric random variable with parameter p if its PMF is given by the equation below. The random experiment for this distribution consists of performing repeated independent “Bernoulli” trials until a given outcome (e.g. heads for a coin toss) occurs. The range of X is 1,2,3,… and describes the number of trials before the specified outcome. Here, 0 < p < 1. PX(k) tends to decline as k grows larger since it is more and more likely the specified outcome will have occurred with more trials.
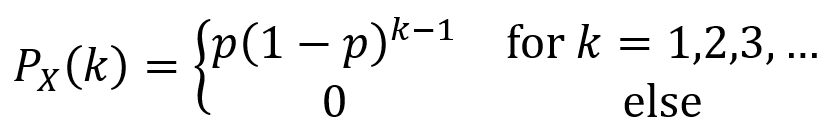
Binomial distribution: a random variable X is a binomial random variable with parameters n and p if its PMF is given by the first equation below. The random experiment for this distribution consists of repeatedly (n times) performing some process which has a probability P(H) = p of yielding an outcome H, then defining X as the total number of times H is observed. Here, 0 < p < 1. Note that the binomial coefficient “n choose k” is given by the second equation below.

Poisson distribution: a random variable X is a Poisson random variable with parameter λ if its range is RX = {0,1,2,3,…} and its PMF is given by the equation below. Poisson distributions are used to describe the count of random occurrences that happen within a certain interval of time or space. For a Poisson distribution, one asks the probability that k events occur will occur within a specified interval wherein an average of λ events are known to happen.

Cumulative distribution functions
The cumulative distribution function (CDF) for a random variable X is defined by the equation below. Note that the CDF is a function over the entire real line, so it must be able to produce an output for any real input value x. An advantage of CDFs is that they can be defined for any type of random variable (discrete, continuous, or mixed).

One can also use CDFs as follows to find the probability that X produces an outcome between two values a and b for which a ≤ b. Note that it is important to pay attention to when the “<” and “≤” symbols can be used.
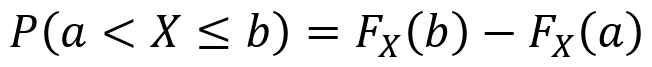
Expectation
The expectation value E of a discrete random variable X is its average or mean, also described as a weighted average of the values in its range. The expectation value formula is given by the first equation below, along with various equivalent notational options. Here, the range RX of the random variable X can be finite or countably infinite. An important property of expectation is that it is linear. The linearity of expectation is described by the second equation below.
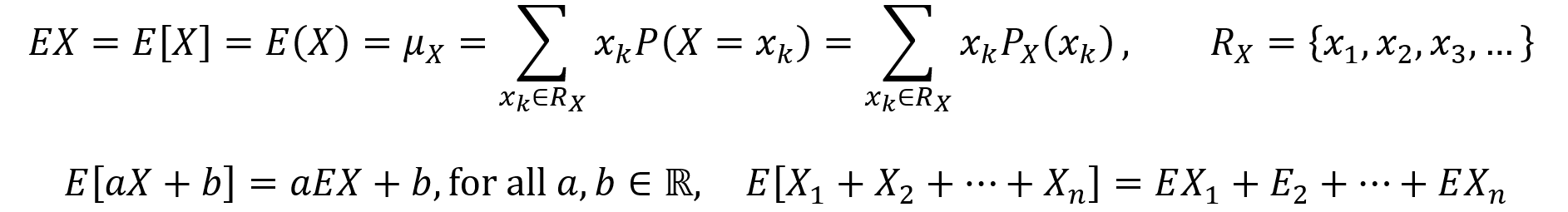
Functions of random variables
If X is a random variable and there is a function Y = g(X), then Y is also a random variable. The range of Y is RY = {g(x) | x ∈ RX} and the PMF of Y is PY(g(X) = y). A convenient way to find the expected value of E[Y] is given by the law of the unconscious statistician (LOTUS), which is described in the equation below. One can also find the PMF of Y and then use the expectation value formula, but this is usually more difficult than using LOTUS.

Variance
The variance of a random variable with mean µX is a measure of the amount of spread of its distribution. It can be found using either of the following first two equations. Some useful properties of variance are given by the second two equations.

Since variance of X has different units than X itself (variance units are the square of the units of X), standard deviation is often used as an alternative measure of distributional spread. Standard deviation is simply the square root of the variance.

Continuous random variables and probability density functions
A random variable X with a CDF of FX(x) is continuous if FX(x) is a continuous function for all x ∈ ℝ.
CDFs work properly for continuous random variables. However, PMFs are invalid for continuous random variables because P(X = x) = 0 for x ∈ ℝ when each P(X = x) corresponds to an infinitesimal slice of the probability. A different tool is needed. Specifically, the probability density function or PDF is utilized. The PDF fX(x) for a random variable X with a continuous CDF FX(x) for all x ∈ ℝ is defined by the first equation below. PDF properties are given by the rest of the equations below.

Expected value and variance for continuous random variables
The expected value and LOTUS for continuous random variables are analogous to those in the discrete case, but with integrals instead of summations. For a continuous random variable X, the expectation value E[X] is given by the first equation below and the LOTUS is given by the second equation below.
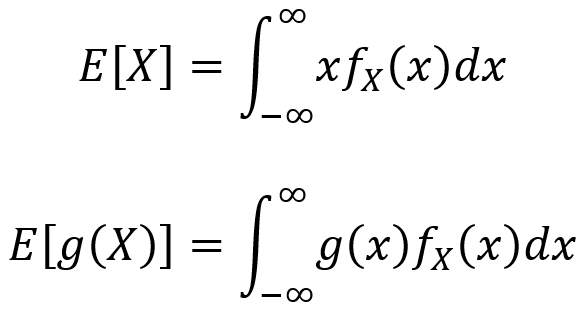
Likewise, the variance for a continuous random variable X takes on the following equivalent forms given by the first two equations below. A useful property of continuous variance is given by the third equation below.

Finding PDFs of functions of continuous random variables
The most straightforward way to find the PDF of a continuous random variable is to start with the CDF and take its derivative (assuming the CDF is continuous).
Another way to find the PDF of a function of a continuous random variable Y = g(X) is to use the method of transformations. The simplest form of this approach requires g to be strictly increasing or strictly decreasing (a monotonic function). However, one can generalize the method of transformations by partitioning the domain RX into n intervals (where n is finite) which each are monotonic and differentiable, even if the function is not monotonic as a whole. This approach for finding the PDF is then given by the following equation.

Special continuous distributions
Uniform distribution: a continuous random variable X has a uniform distribution over the interval [a,b] if its PDF is given as follows.

Exponential distribution: a continuous random variable X has an exponential distribution with parameter λ > 0 if its PDF is given as follows.

Standard normal (standard Gaussian) distribution: a continuous random variable Z has a standard normal distribution if its PDF is given as follows (for all z ∈ ℝ). It should be noted that the standard normal distribution can be scaled and shifted to obtain other normal distributions as will be described after this discussion on the standard normal distribution.

The standard normal distribution has E[Z] = 0 and Var(Z) = 1. The CDF Φ(x) of the standard normal distribution is given by the following equation.
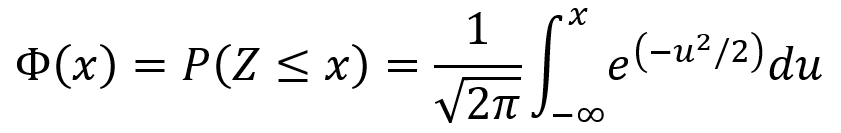
Normal (Gaussian) distributions and normal random variables: if Z is a standard normal random variable and X = σZ + µ, then X is a normal random variable with mean µ and variance σ2. That is, X ~ N(µ,σ2). For any normal variable X with mean µ and variance σ2, the PDF is given by the first equation below and the CDF is given by the second equation below. Additionally, the probability P(a < X ≤ b) is given by the third equation below.

An important property of the normal distribution is that a linear transformation of a normal random variable is itself a normal random variable. This is described by the following theorem.
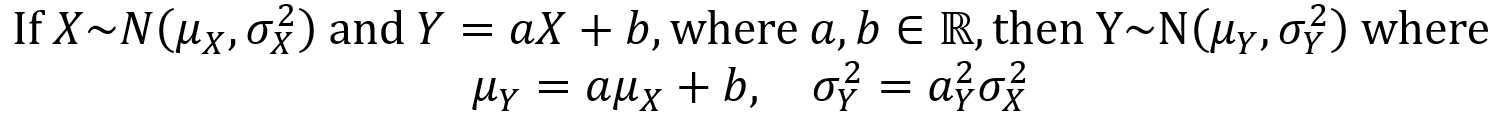
Gamma function and the Gamma distribution: the gamma distribution makes use of the gamma function, which is defined for natural numbers n = {1,2,3,…} by the first equation below and more generally for positive real numbers by the second equation below.

Some properties of the gamma function for any positive real number α > 0 are given by the five equations below.

Now that the definition and properties of the gamma function have been laid out, one can move on to the gamma distribution itself. A continuous random variable X has a gamma distribution over the with parameters α > 0 and λ > 0 if its PDF is given as follows.
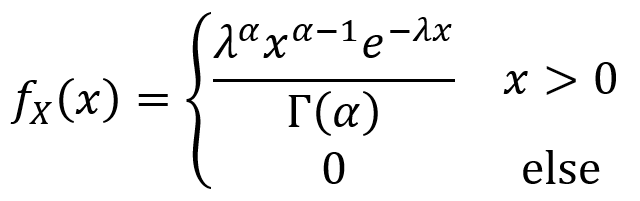
Mixed random variables
When a random variable contains both a continuous part and a discrete part, it is called a mixed random variable. Generally speaking, the CDF of a mixed random variable Y can be written as the sum of a continuous function C(y) and a “staircase” function D(y) as described by the equation below. The plot of FY(y) versus y can have continuous curves, discontinuities between the curves and the discrete parts. The discontinuities sometimes take the form of “jumps”.

To obtain the PDF of a mixed random variable Y, one can integrate the continuous part and turn the discrete part into a sum as described by the following equation. Here, {y1,y2,y3,…} are a set of jump points of D(y). That is, they are the points for which P(Y = yk) > 0. With these mechanics, the PDF over negative infinity to infinity as well as over all jump points yk integrates and sums to 1, ensuring that the axioms of probability are satisfied.
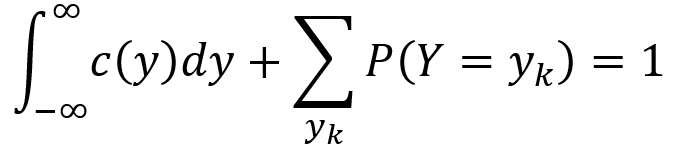
By modifying the equation above, one can obtain the expectation value of Y as shown in the following formula.

Dirac delta function and generalized PDFs
The Dirac delta function is not technically a function but a “generalized function” since it outputs an infinite value for x = 0. It is very useful since it can be used to define a PDF for discrete and mixed random variables. The Dirac Delta function δ(x) and its properties are given by the following equations. Note that u(x) is called the unit step function.
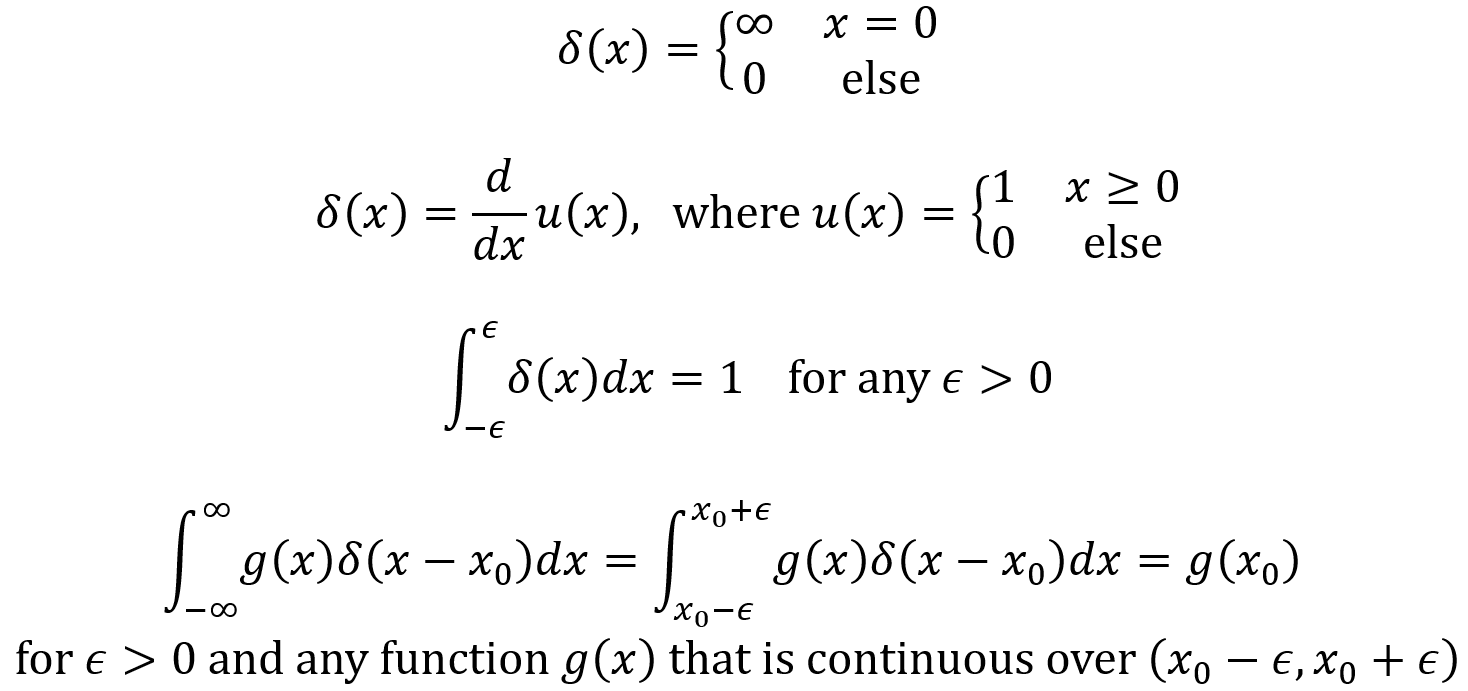
By using the Dirac delta function, one can define a generalized PDF for a discrete random variable as follow. For the random variable X here, the range RX = {x1,x2,x3,…} and the PMF is PX(xk).
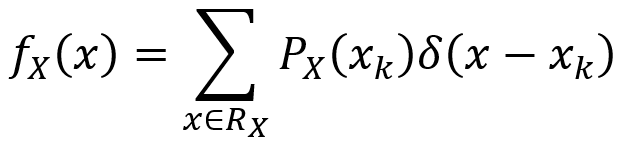
More broadly, the generalized PDF for a mixed random variable X is given by the first equation below. The second equation below describes how this generalized PDF satisfies the axioms of probability since its integral from negative infinity to infinity is 1.
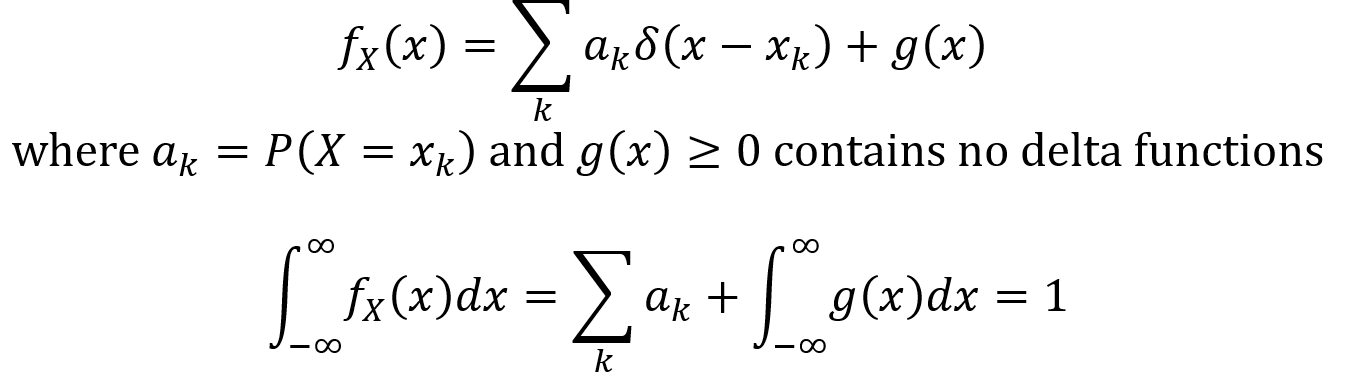

I have long suspected that panpsychism represents the most likely explanation of how consciousness works. My evidence for this claim is laid out below. That said, I am not an expert in philosophy of mind, so take this with a grain of salt. I am certainly open to constructive critiques, questions, and discussion as well!
Supporting points
Organism complexity gradient argument
There appears to be a hierarchy of cognitive complexity ranging roughly down from humans to other primates to other large mammals (e.g. dogs and pigs) to smaller organisms (e.g. mice and rats) to insects to microorganisms to subcellular molecular systems to molecules to atoms to subatomic particles. At what point does consciousness end? There is no clear dividing line between “conscious” and “not conscious”. Thus, some matter may have exceedingly simple cognition, but there is likely not any matter lacking some form of primitive consciousness or experiential qualia.
Lack of real boundaries between brain, body, and environment
Approaching the complexity gradient from a different perspective, one can see that the brain is made of matter and is physically embedded in a body that is also made of matter. Furthermore, the body is physically embedded in an environment made of matter. Where does the “conscious part” end? How does one distinguish between atoms at the edge of the brain which may be thought to participate in conscious processes and atoms at the edge of the pia mater which some may argue do not participate in conscious processes? Furthermore, if a properly configured brain-brain interface was built (think of an electronic cord that physically bridges two people’s brains), it is highly plausible that the two people would experience some of each other’s qualia. Thus, the only real barrier to conscious experiences “spreading” between different organisms seems to be the accuracy of data transfer.
Dualism/supernatural is implied if panpsychism is false
Assume that panpsychism is false. Also assume that the complexity gradient argument holds, implying that there is no clear boundary between conscious and non-conscious material. In this case, where would consciousness exist? It would need to either occupy a sharply defined subset of the universe or need to exist outside of the material universe (i.e. as a supernatural force). But if the gradient argument does not allow us to define a specific subset where consciousness exists, then the only option remaining is for consciousness to exist outside of the material universe. If monist physicalism holds, then panpsychism must be true.
Panpsychism may address the hard problem through physical equivalency
The hard problem confronts many theories of consciousness. In one form of the hard problem, there is the argument that you could have complete mechanistic understanding of how the brain gives rise to a given conscious percept such as the percept of seeing the color red without actually knowing anything about the subjective experience of seeing the color red. However, this may not be true if everything is conscious since it would not be truly possible to have a complete mechanistic understanding of a percept without being physically identical to the matter (e.g. a human) experiencing said percept. Therefore, panpsychism may at least partially address the hard problem of consciousness.
Addressing objections
Reportedly non-conscious parts of brain may actually have qualia
There is neuroscientific evidence that some parts of the brain are active during conscious processing and some parts are not active (subconscious). Yet this is based on the idea that the patient who reports conscious awareness of stimuli represents a unitary entity. What if subcomponents of the brain are instead like different “people”. Perhaps your cerebellum does experience qualia, but just doesn’t transfer most of the data needed to perceive these qualia to your prefrontal cortex. In this way, the conscious parts of the brain would be the ones that have detailed “conversations” with the part of the brain directly involved in the patient’s reporting to the examiner. This goes back to the idea that information transfer may be the only limiting factor in preventing the whole universe from acting as a hive mind. Some parts of the universe do not transfer accurate information to other parts of the universe, but this does not mean that any part of the universe is unconscious.
Anatomy of a rock’s central processing
Some contend that panpsychism is intuitively ludicrous by pointing to the idea that a rock could not possibly be conscious. But consider the following scenario. A rock is illuminated by sunlight on one half of its surface while a shadow from a tree covers the other half. The surface of the rock acts a sensory organ. The rate of diffusion of heat through the rock is governed by factors like the shapes of dense granules packed into the rock’s interior and the composition of the different parts of the rock. The interior of the rock thus acts as a cognitive processor. When the heat comes out from the shadowed side of the rock, different parts of the surface will emit heat at different rates due to the processing that happened inside the rock. The shadowed side of the rock thus acts as a motor output. Certainly, the rock may not have a very accurate model of the world or a system for remembering, predicting, and reflecting. The rock is thus unlikely to have much for self-awareness. Yet it seems plausible that the rock still experiences some form of primitive and noisy qualia. Thinking of the rock in this way makes panpsychism seem less ludicrous.

Illustration of “sensory, integrative/cognitive, and motor” processes that may occur within a rock.

Science Fiction
1. Relinquish / Metamorph (365tomorrows)
2. Queen of the Universe (Aphelion)
3. Honeybee and the Blot (Theme of Absence)
4. Events after the life of Edgar (The Centropic Oracle)
5. Mahabbah (After Dinner Conversation) [purchase required]
6. Oddballs (Across The Margin)
7. The Incandescence of Her Simulacrum (Zooscape)
8. Honeybee and the Blot Reprint (Metastellar)
9. Le Saga Electrik (All Worlds Wayfarer) [purchase required, see reprint for free version]
10. Le Saga Electrik Reprint (Altered Reality Magazine)
11. The Gardener’s Dilemma (Silver Blade Magazine) [try this Wayback Machine link]
12. La biblioteca de las estrellas (White Cat Publications)
13. The First Seed on Mars (Stupefying Stories)
14. The Stairway to Firefly Heaven (Allegory) [no longer available, so here is a local link]
Speculative Poetry
1. The Sonata Machine (Andromeda Spaceways Magazine) [see pages 89-90]
2. Neuraweb (Abyss & Apex Magazine)
3. Nueva Shikaga (Altered Reality Magazine) [no longer available]
4. Gorgeous Geometries (Altered Reality Magazine) [no longer available]
5. cyberjinn (Altered Reality Magazine) [no longer available]
6. Glimmerglimpse (Mithila Review)
7. Electrocologies (Mithila Review)
8. Foreversong (Silver Blade Magazine) [no longer available]
