PDF version: An Introduction to Ebolavirus Biology – Logan Thrasher Collins
I wrote this educational primer as a fun exploration of a topic not related to my current research. While such knowledge may be useful in the event of some future ebolavirus epidemic, it is mostly just an exercise in curiosity and intellectual enrichment. I hope that you too enjoy learning about this fascinating (but scary!) virus as you browse my writeup. Also, if you’re an ebolavirus expert with concepts, edits, and/or ideas to offer, feel free to reach out with your additional insights! Shoutout: I’d like to give a special shoutout/thanks to Jain et al. (reference 4) and Bodmer et al. (reference 2). I used their papers extensively throughout the creation of writeup!
Genome
The ebolavirus genome consists of an 18.9 kb negative-sense single-stranded RNA (ssRNA) which encodes seven genes.1,2 Each gene is flanked by a 3’ and 5’ untranslated region (3’UTR and 5’UTR) which contain start and end signals. The start signals have the consensus sequence of 3’-CUNCUUCUAAUU-5’ and the end signals have the consensus sequence 3’-UAAUUC(U)5/6-5’. Since 3’UAAUU-5’ is found in both the start and end signals, they can overlap and (for most types of ebolavirus) do so at the junctions between the VP35-VP40, GP-VP30, and VP24-L genes. The rest of the genes have intergenic regions with non-overlapping start and end signals between them.
The 5’ and 3’ ends of the genome contain elements called the 5’ trailer and 3’ leader. The 5’ trailer contains parts of the antigenomic replication promoter and the 3’ leader contains parts of the genomic replication promoter. There is also a second genomic replication promoter in the NP untranslated region. Genomic replication promoters initiate RNA-dependent RNA polymerase (RdRP) replication of the negative-sense ssRNA genome while antigenomic replication promoters initiate replication of the positive-sense copy version of the ssRNA genome.
In total, the ebolavirus genome encodes seven proteins.1 The seven proteins encoded by the ebolavirus genome include NP (nucleoprotein), VP24 (membrane-associated protein interfering with interferon signaling), VP30 and VP35 (polymerase matrix protein acting as interferon antagonist), L (the RdRP for replication), VP40 (matrix protein), and GP (glycoprotein).1,2 The proteins will be discussed with more detail in the next section.
The GP RNA itself undergoes mRNA editing, so the GP can take three different forms.2,3 The unedited GP mRNA (~80% of transcripts) encodes a precursor of soluble glycoprotein or sGP. The edited GP0 mRNA (~20% of transcripts)4,5 arises from viral polymerase stuttering at a slippage region sequence of seven consecutive uridines, which leads to addition of an adenosine and a frameshift allowing expression of GP1,2. VP30 may help facilitate resolution of a stem loop involved in the stuttering of the viral polymerase.6 Finally, sometimes either two adenosines are added or one adenosine is omitted from the mRNA (5% of transcripts), leading instead to expression of a small soluble GP precursor protein (ssGP).2,3

Structure
At a glance, ebolavirus consists of its ssRNA genome, a nucleocapsid and accessory proteins, and an envelope bearing its glycoproteins. The NP adopts a helical structure when complexed with the ssRNA genome, forming the nucleocapsid.7 VP35 and VP24 associate with the surface of the NP-RNA complex. VP40 forms the matrix between the envelope and the nucleocapsid. VP30 also binds the nucleocapsid and is important for transcription initiation.4 GP is a transmembrane protein which plays roles in cellular attachment and transduction.

NP
NP’s main function is to encapsidate the ssRNA genome, forming a helical ssRNA-NP complex (the nucleocapsid).2,8 The NPs form a left-handed helix with 24 subunits per turn. Each NP subunit binds six nucleotides of ssRNA via a positively charged cleft on the outside of the NP helix. The NP forms the core of a repeating asymmetric unit consisting of two NPs associated with two oppositely-oriented VP24 proteins, one of which in turn associates with a VP35 protein.
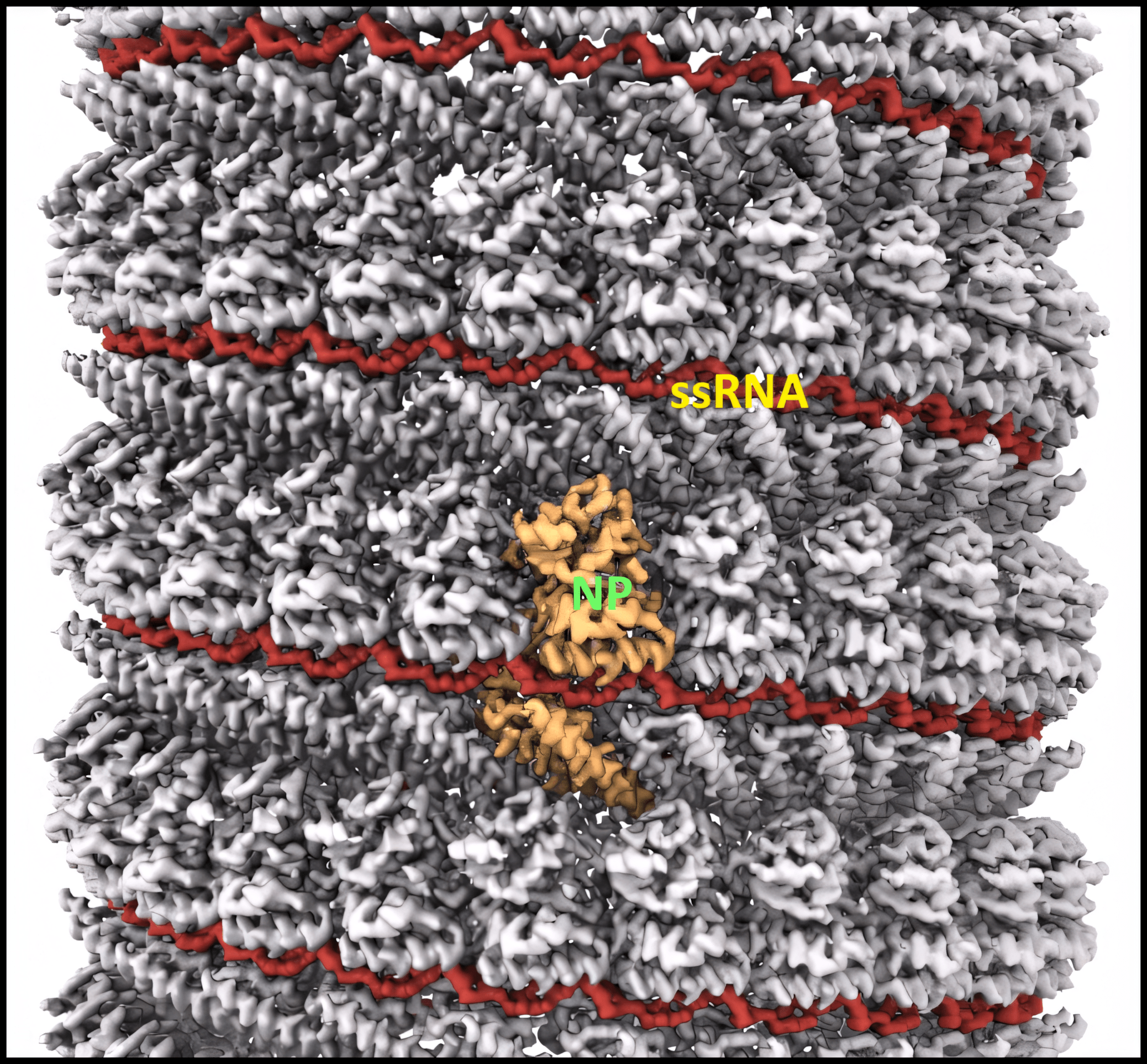
Image adapted from reference 8 (Sugita et al.)
In the cryo-EM structure8 above, the nucleocapsid helix of NP and ssRNA is displayed. VP24 and VP35 are not shown, though they also associate with the nucleocapsid.
VP24
VP24’s interactions and association with NP are required for nucleocapsid formation as well as for helping package the nucleocapsid into virions.4 It is involved in the initiation of viral budding. VP24 additionally inhibits the host cell’s immune responses. It inhibits IFN responses by blocking p38 phosphorylation, which inhibits the p38 MAPK pathway. It also can block NF-κB activation, precluding multiple downstream IFN gene expression pathways. VP24 can inhibit nuclear translocation of the phosphorylated transcription factor STAT1 by interacting with importins of the NPI-1 subfamily of importin-α.
VP35
VP35 is a tetrameric protein which plays a structural role in ebolavirus by associating with the surface of the nucleocapsid. It furthermore acts as a polymerase cofactor which bridges the NP-RNA complex with L (the polymerase) during replication.7 It has helicase and NTPase activities, which indicate that it may unwind RNA helices and hydrolyze NTPs to facilitate transcription and replication.4 VP35 also helps facilitate genome packaging and nucleocapsid assembly.
In addition, VP35 inhibits host cell immune responses.4 It interferes with the dimerization, phosphorylation, and nuclear localization of interferon regulatory factor 3 (IRF-3). It accomplishes this by preventing proteins TBK-1 and IKKε from interacting with IRF-3. Under normal circumstances, phosphorylation and dimerization of IRF-3 causes it to translocate to the nucleus and induce transcription of IFNα, IFNβ, and other genes. VP35 furthermore suppresses interferon transcription by enhancing SUMOylation of IRF-7 via interaction with PIAS1 (a type of SUMO ligase). VP35 also blocks PACT (which prevents activation of PACT-induced RIG-I ATPase) as well as inactivating PKR.
VP30
VP30 forms a hexamer composed of three dimers.4 It is required for RNA transcription initiation. It should be noted that VP30 has a disordered arginine-rich region in the middle of its sequence which interacts with the viral RNA. VP30 also interacts with NP, an interaction which must occur at a certain threshold level for optimal transcriptional activity. VP30 binds zinc, an essential capability for viral transcription initiation.
For transcription to occur, VP30 must either exhibit no phosphorylation (on serines and threonines) or have only partial phosphorylation along with constant phosphorylation-dephosphorylation activity. Partial phosphorylation is only acceptable at some stages of viral replication. By contrast, when it is phosphorylated, VP30 binds NP more robustly. This allows it to tightly associate with the nucleocapsid as new ebolavirus particles are synthesized.
VP40 (matrix protein)
Ebolavirus VP40 is abundantly expressed and associates with the plasma membrane of the host cell, where it facilitates viral assembly and budding.4 It contains two late budding domains (L-domains) of four amino acids each: PTAP and PPEY. The PTAP domain interacts with tumor susceptibility gene 101 protein (tsg101), which recruits VP40 to lipid raft domains on the plasma membrane. PPEY interacts with ubiquitin ligase Nedd4 and ubiquitin ligase ITCH E3, causing ubiquitination of the matrix proteins in certain ways, a requirement for budding.
VP40 can form dimers, hexamers, filaments, and octamers. Dimerization of VP40 is essential for binding to Sec24c and trafficking to the plasma membrane. Sec24c is a component of the coat protein complex II (COPII) which facilitates formation of transport vesicles traveling from the endoplasmic reticulum to the Golgi apparatus, enabling eventual transport to the plasma membrane.9 Dimers can assemble into filaments via VP40’s C-terminal domain residues, which is crucial for matrix assembly and budding.4
VP40 contains a C-terminal domain with a hydrophobic interface which penetrates the plasma membrane to anchor the matrix protein and facilitate assembly and budding.10 Interestingly, VP40 has been shown to selectively anchor onto the plasma membrane via interactions with the enriched anionic phospholipids like phosphatidylserine found in the plasma membrane. At the membrane, the dimers assemble into linear hexamers which are also important for assembly and budding. VP40 can additionally form octameric rings which are essential for VP40-ssRNA binding. Oligomers of VP40 have also been implicated as inhibitory regulators of viral transcription.11
L protein
The L protein is the RNA-dependent RNA polymerase (RdRP) of the ebolavirus.4 It is a fairly large (2212 amino acids) protein consisting of five domains: (i) the RdRP domain which facilitates transcription and replication and polyadenylation, (ii) the capping domain which has polyribonucleotidyl transferase activity, (iii) a connector domain, (iv) a methyltransferase domain, and (v) a small C-terminal domain. The capping domain transfers a GDP to the 5’ phosphate of the viral mRNA. The methyltransferase then methylates the first nucleotide at the 2’-O position and the guanosine cap at the N-7 position. The small C-terminal domain plays a role in recruiting RNAs before methylation.
Additionally, the first 450 amino acids contain a homo-oligomerization domain which overlaps with the RdRP domain. The first 380 amino acids furthermore contain a domain for interaction with the VP35 protein, allowing localization of the L protein into viral inclusion bodies during assembly.
GP
GP (glycoprotein) is a fusogenic transmembrane protein.4 It has a cathepsin binding site which is cleaved inside the endosome as a step in viral infection. It is also post-translationally cleaved by furin from its precursor GP0 to make GP1 and GP2 subunits, which remain linked by disulfide bonds. Three GP1,2 complexes associate to form the trimeric GP that is displayed on the ebolavirus envelope surface.5
GP1 mediates attachment to host cell receptors via its receptor binding domain (RBD).4 There is a heavily glycosylated mucin-like domain (MLD) in GP1, which can stimulate host dendritic cells by activating their MAPK and NF-κB pathways. GP1 furthermore contains another important heavily glycosylated domain called the glycan cap (though it is in the middle of the GP1 sequence).12
GP2 facilitates fusion of viral envelopes with host cell membranes. It does this by inserting a hydrophobic loop domain into the endosomal membrane, bringing the envelope into close contact with said endosomal membrane.4 GP2 also contains a transmembrane anchor domain to help tether the GP to the envelope. GP2 furthermore can inhibit cellular antiviral responses. Firstly, it interferes with tetherin activity (tetherins are host cell proteins that aim to prevent viral budding by “tethering” the virus). It also interferes with NF-κB signaling pathways. GP2 can additionally trigger lymphocyte apoptosis and cytokine dysregulation via an immunosuppressive C-terminal motif.
GP is subject to heavy post-translational glycosylation, protecting the protein against host antibodies.4 GP1 contains 95 glycosylation sites and GP2 contains an additional 2 known glycosylation sites. In particular, MLD is highly glycosylated, allowing it to mask cell-surface proteins like MHC-I (thus inhibiting CD8+ T cell responses).
GP1,2 can be cleaved away from the viral envelope by the host enzyme TACE (TNF-α converting enzyme), leading to shed GP.4,13 The shed GP can sequester antibodies, acting as an immunological decoy. Shed GP furthermore contributes to triggering various inflammatory cytokines.
GP1,2 expression makes up only 20% of the total expressed protein from the GP gene.4,5 The other 80% consists of soluble secreted glycoprotein (sGP), Δ peptide, and small soluble secreted glycoprotein (ssGP). Both the GP1,2 and the ssGP are transcribed only when different ribosomal stuttering events occur during transcription as described earlier (in the genome section).
The sGP is a secreted protein which may serve as an immunological decoy which (as with shed GP) binds antibodies and thereby reduces the available antibodies that can bind to the virus itself.4,5 It has 7 glycosylation sites. In addition, sGP might inhibit inflammatory cytokines and chemokines, further helping the virus evade immunological responses.4
A small C-terminal region of sGP can be cleaved off to form Δ peptide.4,5 The Δ peptide also acts as an immunological decoy. Δ peptide can inhibit entry of ebolavirus into certain cells, preventing superinfection. In addition, Δ peptide may act as a viroporin, forming pores in mammalian cells.
The ssGP consists of an N-terminal region of 295 amino acids which are identical to GP0 and sGP and a C-terminal region of 3 amino acids which are distinct. It is secreted as a disulfide bonded homodimer which undergoes glycosylation. Its function remains unknown.4

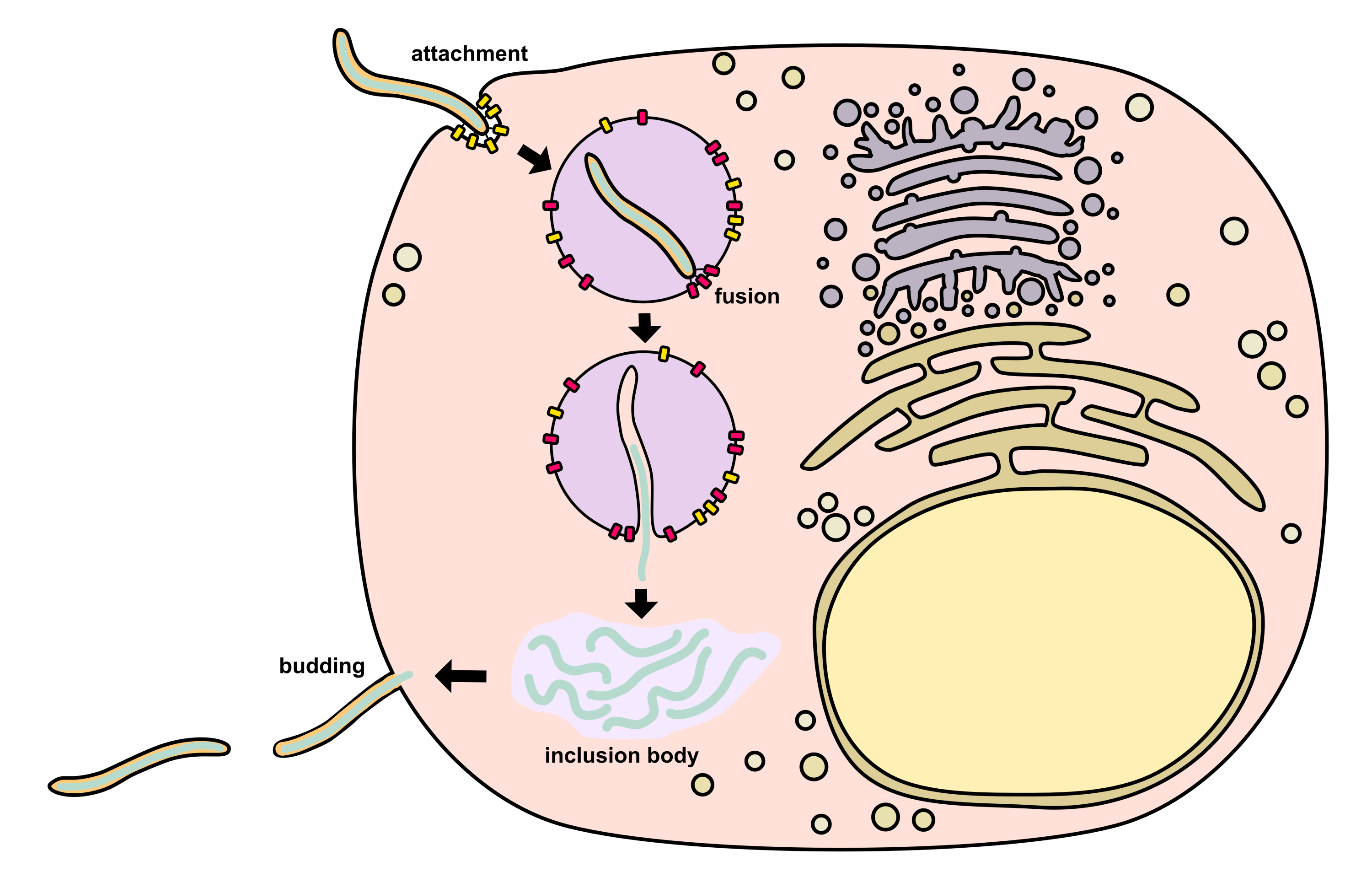
Life Cycle
Attachment
Ebolavirus begins its life cycle by leveraging GP1,2 to attach to host cell receptors.2 There are three known mechanisms for attachment including (i) binding of C-type lectins, (ii) interaction with phosphatidylserine-binding receptors, and (iii) antibody-dependent enhancement.
C-type lectins bind the GP’s glycans found on the MLD as well as the glycan cap.2 Such lectins are mainly expressed on antigen presenting cells (dendritic cells, monocytes, macrophages, etc.) which are a primary target cell of ebolaviruses. However, they are not required or sufficient for entry, so they act as accessory receptors.
During budding, ebolavirus incorporates the host scramblase XKR8 into its envelope (via interactions with GP1,2),2 which randomly swaps phospholipids between inner and outer membrane leaflets. It should be noted that other scramblases like TMEM16F might also be used by the virus in the same way. The scramblases expose phosphatidylserine on the envelope’s surface (phosphatidylserine is normally found on the inner leaflet rather than the outer leaflet). As a result, phosphatidylserine receptors (TIM-1, TIM-4, Axl, and Mer) on the host cell membrane can bind the phosphatidylserine on the viral envelope. Since exposure of phosphatidylserine is normally used by the host to induce phagocytosis of apoptotic cell debris, the presence of phosphatidylserine on the ebolavirus envelope targets it for uptake into phagocytes. This is called “apoptotic mimicry”.
Antibody-dependent enhancement is when anti-ebolavirus antibodies bind to the virus and immune cell Fc receptors bind the antibodies.2 Complement factor C1q can also bind the ebolavirus-antibody complexes and attach virions to immune cell surfaces. Though these pathways normally facilitate clearing of viruses by endocytic uptake and degradation, ebolavirus may leverage the process for infection instead.
Endosomal trafficking and fusion
After binding the cell surface, the ebolavirus is endocytosed via macropinocytosis, preferentially near host cell membrane lipid rafts.2 Virions are trafficked from the early endosome to the late endosome. In the late endosome, the GP’s MLD and glycan cap are cleaved off by cathepsin B, cathepsin L, and/or other host cell proteases. This allows the GP to bind the intracellular receptor NPC1 (Niemann–Pick C1), which is found on the inner surface of the endosome. Low pH in the endosome causes acidification in the virus, which triggers disassociation of the VP40 matrix protein from the envelope, granting the virus more flexibility. It is thought that this flexibility may represent an additional prerequisite for fusion. Finally, the GP experiences a conformal change that causes insertion of GP2’s hydrophobic loop domain into the endosomal membrane, facilitating fusion with the envelope (a process dependent on certain pH and Ca2+ levels). After fusion, the nucleocapsid is released into the cytosol.
Transcription
Condensed nucleocapsids in the cytosol next begin RNA synthesis.2 To do this, they use a ribonucleoprotein complex consisting of L, NP, VP35, and VP30. Primary transcription relies on proteins from the incoming virion while secondary transcription can also utilize proteins newly produced inside the host cell. L (along with its VP35 cofactor) catalyzes RNA polymerization as well as methylation (capping) of viral mRNAs as discussed earlier.
Cytosolic transcription initiated by the polymerase complex is assisted by VP30.2 L starts at a site at the 3’ end of the genome and scans for the start signal of the first gene, which is the NP gene. The mRNA’s polyadenylation is triggered via polymerase slippage at the poly-uridine end signals which were discussed previously. L continues scanning the genome for the next start signal. It should be noted that scanning can occur in both directions along the genome. If the polymerase disassociates from the genome during scanning, it must return to the 3’ end to reinitiate. Because of this, genes close to the 3’ end of the genome are transcribed at a higher level than genes towards the 5’ end, a transcriptional gradient which might have functional significance.
Primary transcription occurs within 1-2 hours after infection.2 After ~10 hours post-infection, ebolavirus causes the formation of cytosolic inclusion bodies that serve to facilitate secondary transcription and genome replication. These inclusion bodies are rich in L, NP, VP35, and VP30 as well as VP24, VP40, and certain host proteins such as CAD, STAU1, SMYD3, RBBP6, PEG10, hnRNP L, and RUVBL1. Ebolavirus inclusion bodies occur as membraneless phase-separated condensates driven by NP oligomerization interactions.
Viral mRNAs are exported from inclusion bodies by recruiting host NFX1 (nuclear RNA export factor 1).2 NFX1 binds mRNAs within inclusion bodies and transports them out into the cytosol, where translation can occur. It has been shown that hypusinated eIF5A (eukaryotic initiation 5A) is required for viral mRNA translation. Note that hypusination is a post-translational modification where a lysine in eIF5A is converted to a non-canonical amino acid called hypusine.14 In addition, ADAR1 (adenosine deaminase acting on RNA 1) edits 3’ untranslated regions within viral mRNAs and thus alters some of their negative regulatory elements to no longer downregulate translation.2
The transition from transcription to replication is thought to involve VP30 phosphorylation.2 Non-phosphorylated VP30 associates more strongly with the L-VP35 complex than its phosphorylated form. Phosphorylation of VP30 (and its lower affinity for L-VP35 in this form) may shift the focus of L-VP35 towards replication. When VP30 is phosphorylated, it also interacts more strongly with NP, which helps VP30 incorporate itself into new virions. That said, this process is not fully understood. Cellular kinases (SRPK1 and SRPK2) and phosphatases (PP2A-B56 and PP1) are sequestered into viral inclusion bodies to facilitate VP30 phosphorylation and dephosphorylation.
Replication
Only L, VP35, and NP are needed for ebolavirus replication (unlike transcription, which also needs VP30).2 For replication, the genome is copied into an antigenome. Replication is initiated at the first C in the genome, which is actually position 2 in the sequence. As a result, the copies initially lack the 3’-terminal nucleotide. To fix this, it is believed that the 3’ region of the RNA folds into a hairpin structure which back-primes addition of the missing nucleotide. Both the genome and the antigenome are encapsidated by NP.2 During the replication process, VP35 acts as a chaperone for monomeric NP that has not yet bound RNA. VP24 may cause nucleocapsids to transition from a relaxed state to a more condensed state.
Assembly and budding
After release from inclusion bodies, the nucleocapsids are transported along actin filaments to the plasma membrane where budding takes place.2 GP1,2 reaches the plasma membrane through the secretory pathway since it is a transmembrane protein. VP40 mediates budding by taking over parts of the host’s ESCRT (endosomal sorting complex required for transport) pathway. VP40 has a motif which recruits Tsg101 (an ESCRT-I component) to lipid rafts in the membrane. VP40 also has a motif which interacts with ubiquitin protein ligases (NEDD4, ITCH, WWP1, and SMURF2). These ubiquitin ligases ubiquitinate VP40, which facilitates its activity in budding. VP40 may also induce curvature across membrane phospholipids via its oligomerization. VP40 has a basic patch in its C-terminal domain which interacts with phosphatidylserine, which causes phosphatidylserine to cluster. Indeed, it has been shown that proper matrix layer formation requires phosphatidylserine clustering, so the interaction likely has functional importance.
References
1. Ghosh, S., Saha, A., Samanta, S. & Saha, R. P. Genome structure and genetic diversity in the Ebola virus. Curr. Opin. Pharmacol. 60, 83–90 (2021).
2. Bodmer, B. S., Hoenen, T. & Wendt, L. Molecular insights into the Ebola virus life cycle. Nat. Microbiol. 9, 1417–1426 (2024).
3. Martin, B., Hoenen, T., Canard, B. & Decroly, E. Filovirus proteins for antiviral drug discovery: A structure/function analysis of surface glycoproteins and virus entry. Antiviral Res. 135, 1–14 (2016).
4. Jain, S., Martynova, E., Rizvanov, A., Khaiboullina, S. & Baranwal, M. Structural and Functional Aspects of Ebola Virus Proteins. Pathogens vol. 10 at https://doi.org/10.3390/pathogens10101330 (2021).
5. Lee, J. E. & Saphire, E. O. Ebolavirus Glycoprotein Structure and Mechanism of Entry. Future Virol. 4, 621–635 (2009).
6. Mehedi, M. et al. Ebola Virus RNA Editing Depends on the Primary Editing Site Sequence and an Upstream Secondary Structure. PLOS Pathog. 9, e1003677 (2013).
7. Fujita-Fujiharu, Y. et al. Structural basis for Ebola virus nucleocapsid assembly and function regulated by VP24. Nat. Commun. 16, 2171 (2025).
8. Sugita, Y., Matsunami, H., Kawaoka, Y., Noda, T. & Wolf, M. Cryo-EM structure of the Ebola virus nucleoprotein–RNA complex at 3.6 Å resolution. Nature 563, 137–140 (2018).
9. Mancias, J. D. & Goldberg, J. Structural basis of cargo membrane protein discrimination by the human COPII coat machinery. EMBO J. 27, 2918–2928 (2008).
10. Adu-Gyamfi, E. et al. The Ebola Virus Matrix Protein Penetrates into the Plasma Membrane. J. Biol. Chem. 288, 5779–5789 (2013).
11. T., H. et al. Oligomerization of Ebola Virus VP40 Is Essential for Particle Morphogenesis and Regulation of Viral Transcription. J. Virol. 84, 7053–7063 (2010).
12. Peng, W. et al. Glycan shield of the ebolavirus envelope glycoprotein GP. Commun. Biol. 5, 785 (2022).
13. Ning, Y.-J., Deng, F., Hu, Z. & Wang, H. The roles of ebolavirus glycoproteins in viral pathogenesis. Virol. Sin. 32, 3–15 (2017).
14. McKenna, S. The first step of hypusination. Nat. Chem. Biol. 19, 664 (2023).